 Scoop has an Ethical Paywall
Scoop has an Ethical Paywall


A Proposed Origin For SARS-CoV-2 And The COVID-19 Pandemic
by Jonathan Latham, PhD and Allison Wilson, PhD
In all the discussions of the origin of the COVID-19 pandemic, enormous scientific attention has been paid to the molecular character of the SARS-CoV-2 virus, including its novel genome sequence in comparison with its near relatives. In stark contrast, virtually no attention has been paid to the physical provenance of those nearest genetic relatives, its presumptive ancestors, which are two viral sequences named BtCoV/4991 and RaTG13.
This neglect is surprising because their provenance is more than interesting. BtCoV/4991 and RaTG13 were collected from a mineshaft in Yunnan province, China, in 2012/2013 by researchers from the lab of Zheng-li Shi at the Wuhan Institute of Virology (WIV). Very shortly before, in the spring of 2012, six miners working in the mine had contracted a mysterious illness and three of them had died (Wu et al., 2014). The specifics of this mystery disease have been virtually forgotten; however, they are described in a Chinese Master’s thesis written in 2013 by a doctor who supervised their treatment.
We arranged to have this Master’s thesis translated into English. The evidence it contains has led us to reconsider everything we thought we knew about the origins of the COVID-19 pandemic. It has also led us to theorise a plausible route by which an apparently isolated disease outbreak in a mine in 2012 led to a global pandemic in 2019.
The origin of SARS-CoV-2 that we propose below is based on the case histories of these miners and their hospital treatment. This simple theory accounts for all the key features of the novel SARS-CoV-2 virus, including ones that have puzzled virologists since the outbreak began.
The theory can account for the origin of the polybasic furin cleavage site, which is a region of the viral spike protein that makes it susceptible to cleavage by the host enzyme furin and which greatly enhances viral spread in the body. This furin site is novel to SARS-CoV-2 compared to its near relatives (Coutard, et al., 2020). The theory also explains the exceptional affinity of the virus spike protein for human receptors, which has also surprised virologists (Letko et al., 2020; Piplani et al, 2020; Wrapp et al., 2020; Walls et al., 2020). The theory further explains why the virus has barely evolved since the pandemic began, which is also a deeply puzzling aspect of a virus supposedly new to humans (Zhan et al., 2020; van Dorp et al., 2020; Chaw et al., 2020). Lastly, the theory neatly explains why SARS-CoV-2 targets the lungs, which is unusual for a coronavirus (Huang et al., 2020).
We do not propose a specifically genetically engineered or biowarfare origin for the virus but the theory does propose an essential causative role in the pandemic for scientific research carried out by the laboratory of Zheng-li Shi at the WIV; thus also explaining Wuhan as the location of the epicentre.
Why has the provenance of RaTG13 and BtCoV/4991 been ignored?
The apparent origin of the COVID-19 pandemic is the city of Wuhan in Hubei province, China. Wuhan is also home to the world’s leading research centre for bat coronaviruses. There are two virology labs in the city, both have either collected bat coronaviruses or researched them in the recent past. The Shi lab, which collected BtCoV/4991 and RaTG13, recently received grants to evaluate by experiment the potential for pandemic pathogenicity of the novel bat coronaviruses they collected from the wild.
To add to these suggestive data points, there is a long history of accidents, disease outbreaks, and even pandemics resulting from lab accidents with viruses (Furmanski, 2014; Weiss et al., 2015). For these and other reasons, summarised in our article The Case is Building that COVID-19 Had a Lab Origin, we (a virologist and a geneticist) and others have concluded that a lab outbreak is a credible thesis. Certainly, a lab origin has at least as much circumstantial evidence to support it as does any natural zoonotic origin theory (Piplani et al., 2020; Segreto and Deigin, 2020; Zhan et al., 2020).
The media, normally so enamoured of controversy, has largely declined even to debate the possibility of a laboratory escape. Many news sites have simply labelled it a conspiracy theory.
The principal reason for media dismissals of the lab origin possibility is a review paper in Nature Medicine (Andersen et al., 2020). Although by Jun 29 2020 this review had almost 700 citations it also has major scientific shortcomings. These flaws are worth understanding in their own right but they are also useful background for understanding the implications of the Master’s thesis.
Andersen et al., a critique
The question of the origin of the COVID-19 pandemic is, in outline, simple. There are two incontrovertible facts. One, the disease is caused by a human viral pathogen, SARS-CoV-2, first identified in Wuhan in December 2019 and whose RNA genome sequence is known. Second, all of its nearest known relatives come from bats. Beyond any reasonable doubt SARS-CoV-2 evolved from an ancestral bat virus. The task the Nature Medicine authors set for themselves was to establish the relative merits of each of the various possible routes (lab vs natural) by which a bat coronavirus might have jumped to humans and in the same process have acquired an unusual furin site and a spike protein having very high affinity for the human ACE2 receptor.
When Andersen et al. outline a natural zoonotic pathway they speculate extensively about how the leap might have occurred. In particular they elaborate on a proposed residence in intermediate animals, likely pangolins. For example, “The presence in pangolins of an RBD [Receptor Binding Domain] very similar to that of SARS-CoV-2 means that we can infer that this was probably in the virus that jumped to humans. This leaves the insertion of [a] polybasic cleavage site to occur during human-to-human transmission.” This viral evolution occurred in “Malayan pangolins illegally imported into Guangdong province”. Even with these speculations there are major gaps in this theory. For example, why is the virus so well adapted to humans? Why Wuhan, which is 1,000 Km from Guangdong? (See map).
The authors provide no such speculations in favour of the lab accident thesis, only speculation against it:
“Finally, the generation of the predicted O-linked glycans is also unlikely to have occurred due to cell-culture passage, as such features suggest the involvement of an immune system.” (italics added).
[Passaging is the deliberate placing of live viruses into cells or organisms to which they are NOT adapted for the purpose of making them adapted, i.e. speeding up their evolution.]
It is also noteworthy that the Andersen authors set a higher hurdle for the lab thesis than the zoonotic thesis. In their account, the lab thesis is required to explain all of the evolution of SARS-CoV-2 from its presumed bat viral ancestor, whereas under their telling of the zoonotic thesis the key step of the addition of the furin site is allowed to happen in humans and is thus effectively unexplained.
A further imbalance is that key information needed to judge the merits of a lab origin theory is missing from their account. As we detailed in our previous article, in their search for SARS-like viruses with zoonotic spillover potential, researchers at the WIV have passaged live bat viruses in monkey and human cells (Wang et al., 2019). They have also performed many recombinant experiments with diverse bat coronaviruses (Ge et al., 2013; Menachery et al., 2015; Hu et al., 2017). Such experiments have generated international concern over the possible creation of potential pandemic viruses (Lipsitch, 2018). As we showed too, the Shi lab had also won a grant to extend that work to whole live animals. They planned “virus infection experiments across a range of cell cultures from different species and humanized mice” with recombinant bat coronaviruses. Yet Andersen et al did not discuss this research at all, except to say:
“Basic research involving passage of bat SARS-CoV-like coronaviruses in cell culture and/or animal models has been ongoing for many years in biosafety level 2 laboratories across the world”
This statement is fundamentally misleading about the kind of research performed at the Shi lab.
A further important oversight by the Andersen authors concerns the history of lab outbreaks of viral pathogens. They write: “there are documented instances of laboratory escapes of SARS-CoV”. This is a rather matter-of-fact allusion to the fact that since 2003 there have been six documented outbreaks of SARS from labs, not all in China, with some leading to fatalities (Furmanski, 2014).
Andersen et al might have also have noted that two major human pandemics are widely accepted to have been caused by lab outbreaks of viral pathogens, H1N1 in 1977 and Venezuelan Equine Encephalitis (summarised in Furmanski, 2014). Andersen could even have noted that literally hundreds of lab accidents with viruses have resulted in near-misses or very localised outbreaks (summarised by Lynn Klotz and Sam Husseini and also Weiss et al., 2015).
Also unmentioned were instances where a lab outbreak of an experimental or engineered virus has been plausibly theorised but remains uninvestigated. For example, the most coherent explanation for the H1N1 variant ‘swine flu’ pandemic of 2009/10 that resulted in a death toll estimated by some as high as 200,000 (Duggal et al., 2016; Simonsen et al. 2013), is that a vaccine was improperly inactivated by its maker (Gibbs et al., 2009). If so, H1N1 emerged from a lab not once but twice.
Given that human and livestock viral outbreaks have frequently come from laboratories and that many scientists have warned of probable lab escapes (Lipsitch and Galvani, 2014), and that the WIV itself has a questionable biosafety record, the Andersen paper is not an even-handed treatment of the possible origins of the COVID-19 virus.
Yet its text expresses some strong opinions: “Our analyses clearly show that SARS-CoV-2 is not a laboratory construct or a purposefully manipulated virus….It is improbable that SARS-CoV-2 emerged through laboratory manipulation of a related SARS-CoV-like coronavirus…..the genetic data irrefutably show that SARS-CoV-2 is not derived from any previously used backbone….the evidence shows that SARS-CoV2 is not a purposefully manipulated virus….we do not believe that any type of laboratory-based scenario is possible.” (Andersen et al., 2020).
It is hard not to conclude that what their paper mostly shows is that Drs. Andersen, Rambaut, Lipkin, Holmes and Garry much prefer the natural zoonotic transfer thesis. Their rhetoric is forthright but the evidence does not support that confidence.
Indeed, since the publication of Andersen et al., important new evidence has emerged that undermines their zoonotic origin theory. On May 26th the Chinese CDC ruled out the Huanan “wet” market in Wuhan as the source of the outbreak. Additionally, new research on pangolins, the favoured intermediate mammal host, suggests they are not a natural reservoir of coronaviruses (Lee et al., 2020; Chan and Zhan, 2020). Furthermore, SARS-CoV-2 was found not to replicate in bat kidney or lung cells (Rhinolophus sinicus), implying that SARS-CoV-2 is not a recently-adapted spill over Chu et al., 2020).
The Mojiang mine and the Master’s thesis
In our own search to resolve the COVID-19 origin question we chose to focus on the provenance of the coronavirus genome sequences BtCoV/4991 and RaTG13, since these are the most closely related sequences to SARS-CoV-2 (98.7% and 96.2% identical respectively). See FIG 1. (reproduced from P. Zhou et al., 2020).
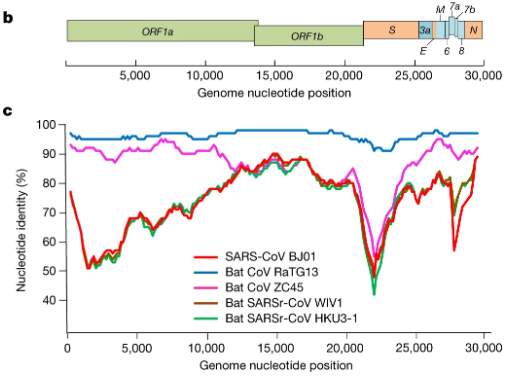
For comparison, the next closest virus to SARS-CoV-2 is RmYN02 (not shown in Fig 1.) (H. Zhou et al., 2020). RmYN02 has an overall similarity to SARS-CoV-2 of 93.2%, making its evolutionary distance from SARS-CoV-2 almost twice as great.
BtCoV/4991 was first described in 2016. It is a 370 nucleotide virus fragment collected from the Mojiang mine in 2013 by the lab of Zeng-li Shi at the WIV (Ge et al., 2016). BtCoV/4991 is 100% identical in sequence to one segment of RaTG13. RaTG13 is a complete viral genome sequence (almost 30,000 nucleotides) that was only published in 2020, after the pandemic began (P. Zhou et al., 2020).
Despite the confusion created by their different names, in a letter obtained by us Zheng-li Shi confirmed to a virology database that BtCoV/4991 and RaTG13 are both from the same bat faecal sample and the same mine. They are thus sequences from the same virus. In the discussion below we will refer primarily to RaTG13 and specify BtCoV/4991 only as necessary.
These specifics are important because it is these samples and their provenance that we believe are ultimately key to unravelling the mystery of the origins of COVID-19.
The story begins in April 2012 when six workers in that same Mojiang mine fell ill from a mystery illness while removing bat faeces. Three of the six subsequently died.
In a March 2020 interview with Scientific American Zeng-li Shi dismissed the significance of these deaths, claiming the miners died of fungal infections. Indeed, no miners or deaths are mentioned in the paper published by the Shi lab documenting the collection of RaTG13 (Ge et al., 2016).
But Shi’s assessment does not tally with any other contemporaneous accounts of the miners and their illness (Rahalkar and Bahulikar, 2020). As these authors have pointed out, Science magazine wrote up part of the incident in 2014 as A New Killer Virus in China?. Science was citing a different team of virologists who found a paramyxovirus in rats from the mine. These virologists told Science they found “no direct relationship between human infection” and their virus. This expedition was later published as the discovery of a new virus called MojV after Mojiang, the locality of the mine (Wu et al., 2014).
What this episode suggests though is that these researchers were looking for a potentially lethal virus and not a lethal fungus. Also searching the Mojiang mine for a virus at around the same time was Canping Huang, the author of a PhD thesis carried out under the supervision of George Gao, the head of the Chinese CDC.
All of this begs the question of why the Shi lab, which has no interest in fungi but a great interest in SARS-like bat coronaviruses, also searched the Mojiang mine for bat viruses on four separate occasions between August 2012 and July 2013, even though the mine is a 1,000 Km from Wuhan (Ge et al., 2016). These collecting trips began while some of the miners were still hospitalised.
Fortunately, a detailed account of the miner’s diagnoses and treatments exists. It is found in a Master’s thesis written in Chinese in May 2013. Its suggestive English title is “The Analysis of 6 Patients with Severe Pneumonia Caused by Unknown viruses“.
The original English version of the abstract implicates a SARS-like coronavirus as the probable causative agent and that the mine “had a lot of bats and bats’ feces”.
The findings of the Master’s thesis
To learn more, especially about the reasonableness of this diagnosis, we arranged to have the whole Master’s thesis translated into English and are here making the translation available. To read it in full see the embedded document below (or download it here).
The six ill miners were admitted to the No. 1. School of Clinical Medicine, Kunming Medical University, in short succession in late April and early May 2012. Kunming is the capital of Yunnan province and 250 Km from Mojiang.
Of the descriptions of the miners and their treatments, which include radiographs and numerous CAT scans, several features stand out:
1) From their admission to the hospital their doctors informed the “medical office” of a potential “outburst of disease” i.e. a potential epidemic outbreak. Thus, the miners were treated for infections and not as if they had inhaled noxious gases or other toxins.
2) The symptoms (on admission) of the six miners were: a) dry cough, b) sputum, c) high fevers, especially shortly before death d) difficulty breathing, e) myalgia (sore limbs). Some patients had hiccoughs and headaches. (See Table 1).
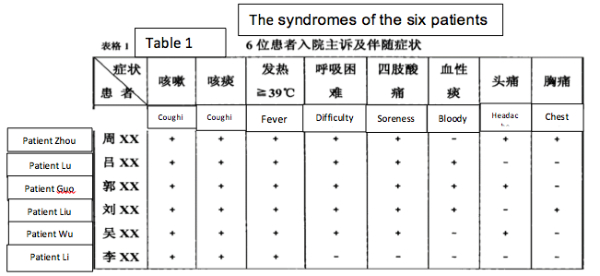
3) Clinical work established that patients 1-4 had low blood oxygen “for sure it was ARDS” (Acute Respiratory Distress Syndrome) and immune damage considered indicative of viral infection. Additionally, a tendency for thrombosis was noted in patients 2 and 4. Symptom severity and mortality were age-related (though from a sample of 6 this must be considered anecdotal).
4) Potential common and rare causes of their symptoms were tested for and mostly eliminated. For patients 3 and 4 these included tests for HIV, Cytomegalovirus, Epstein-Barr Virus (EBV), Japanese encephalitis, haemorrhagic fever, Dengue, Hepatitis B, SARS, and influenza. Of these, only patient 2 tested positive for Hepatitis and EBV.
5) Treatment of the six patients included ventilation (patients 2-4), steroids (all patients), antivirals (all except patient 5), and blood thinners (patients 2 and 4). Antibiotics and antifungal medications were administered to counter what were considered secondary (but significant) co-infections.
6) A small number of remote meetings were held with researchers at other universities. One was with Zhong Nanshan at Sun Yat-Sen University, Guangdong. Zhong is the Chinese hero of the SARS epidemic, a virologist, and arguably the most famous scientist in China.
7) Samples from the miners were later sent to the WIV in Wuhan and to Zhong Nanshan, further confirming that viral disease was strongly suspected. Some miners did test positive for coronavirus (the thesis is unclear on how many).
8) The source of infection was concluded to be Rhinolophus sinicus, a horseshoe bat and the ultimate conclusion of the thesis reads “the unknown virus lead to severe pneumonia could be: The SARS-like-CoV from the Chinese rufous horseshoe bat.” Thus the miners had a coronavirus but it apparently was not SARS itself.
The significance of the Master’s thesis
These findings of the thesis are significant in several ways.
First, in the light of the current coronavirus pandemic it is evident the miners’ symptoms very closely resemble those of COVID-19 (Huang et al, 2020; Tay et al., 2020; M. Zhou et al., 2020). Anyone presenting with them today would immediately be assumed to have COVID-19. Likewise, many of the treatments given to the miners have become standard for COVID-19 (Tay et al., 2020).
Second, the remote meeting with Zhong Nanshan is significant. It implies that the illnesses of the six miners were of high concern and, second, that a SARS-like coronavirus was considered a likely cause.
Third, the abstract, the conclusions, and the general inferences to be made from the Master’s thesis contradict Zheng-li Shi’s assertion that the miners died from a fungal infection. Fungal infection as a potential primary cause was raised but largely discarded.
Fourth, if a SARS-like coronavirus was the source of their illness the implication is that it could directly infect human cells. This would be unusual for a bat coronavirus (Ge et al., 2013). People do sometimes get ill from bat faeces but the standard explanation is histoplasmosis, a fungal infection and not a virus (McKinsey and McKinsey, 2011; Pan et al., 2013).
Fifth, the sampling by the Shi lab found that bat coronaviruses were unusually abundant in the mine (Ge at al., 2016). Among their findings were two betacoronaviruses, one of which was RaTG13 (then known as BtCoV/4991). In the coronavirus world betacoronaviruses are special in that both SARS and MERS, the most deadly of all coronaviruses, are both betacoronaviruses. Thus they are considered to have special pandemic potential, as the concluding sentence of the Shi lab publication which found RaTG13 implied: “special attention should particularly be paid to these lineages of coronaviruses” (Ge at al., 2016). In fact, the Shi and other labs have for years been predicting that bat betacoronaviruses like RaTG13 would go pandemic; so to find RaTG13 where the miners fell ill was a scenario in perfect alignment with their expectations.
The Mojiang miners passaging proposal
How does the Master’s thesis inform the search for a plausible origin of the pandemic?
In our previous article we briefly discussed how the pandemic might have been caused either by a virus collection accident, or through viral passaging, or through genetic engineering and a subsequent lab escape. The genetic engineering possibility deserves attention and is extensively assessed in an important preprint (Segreto and Deigin, 2020).
We do not definitively rule out these possibilities. Indeed it now seems that the Shi lab at the WIV did not forget about RaTG13 but were sequencing its genome in 2017 and 2018. However, we believe that the Master’s thesis indicates a much simpler explanation.
We suggest, first, that inside the miners RaTG13 (or a very similar virus) evolved into SARS-CoV-2, an unusually pathogenic coronavirus highly adapted to humans. Second, that the Shi lab used medical samples taken from the miners and sent to them by Kunming University Hospital for their research. It was this human-adapted virus, now known as SARS-CoV-2, that escaped from the WIV in 2019.
We refer to this COVID-19 origin hypothesis as the Mojiang Miners Passage (MMP) hypothesis.
Passaging is a standard virological technique for adapting viruses to new species, tissues, or cell types. It is normally done by deliberately infecting a new host species or a new host cell type with a high dose of virus. This initial viral infection would ordinarily die out because the host’s immune system vanquishes the ill-adapted virus. But, in passaging, before it does die out a sample is extracted and transferred to a new identical tissue, where viral infection restarts. Done iteratively, this technique (called “serial passaging” or just “passaging”) intensively selects for viruses adapted to the new host or cell type (Herfst et al., 2012).
At first glance RaTG13 is unlikely to have evolved into SARS-CoV-2 since RaTG13 is approximately 1,200 nucleotides (3.8%) different from SARS-CoV-2. Although RaTG13 is the most closely related virus to SARS-CoV-2, this sequence difference still represents a considerable gap. In a media statement evolutionary virologist Edward Holmes has suggested this gap represents 20-50 years of evolution and others have suggested similar figures.
We agree that ordinary rates of evolution would not allow RaTG13 to evolve into SARS-CoV-2 but we also believe that conditions inside the lungs of the miners were far from ordinary. Five major factors specific to the hospitalised miners favoured a very high rate of evolution inside them.
i) When viruses infect new species they typically undergo a period of very rapid evolution because the selection pressure on the invading pathogen is high. The phenomenon of rapid evolution in new hosts is well attested among corona- and other viruses (Makino et al., 1986; Baric et al., 1997; Dudas and Rambaut 2016; Forni et al., 2017).
ii) Judging by their clinical symptoms such as the CT scans, all the miner’s infections were primarily of the lungs. This localisation likely occurred initially because the miners were exerting themselves and therefore inhaling the disturbed bat guano deeply. As miners, they may already have had damaged lung tissues (patient 3 had suspected pneumoconiosis) and/or particulate matter was present that irritated the tissues and may have facilitated initial viral entry.
In contrast, standard coronavirus infections are confined to the throat and upper respiratory tract. They do not normally reach the lungs (Perlman and Netland, 2009). Lungs are far larger tissues by weight (kilos vs grammes) than the upper respiratory tract. There was therefore likely a much larger quantity of virus inside the miners than would be the case in an ordinary coronavirus infection.
Comparing a typical coronavirus respiratory tract infection with the extent of infected lungs in the miners from a purely mathematical point of view indicates the potential scale of this quantitative difference. The human aerodigestive tract is approximately 20cm in length and 5cm in circumference, i.e. approximately 100 cm2 in surface area. The surface area of a human lung ranges from 260,000-680,000 cm2 (Hasleton, 1972). The amount of potentially infected tissue in an average lung is therefore approximately 4500-fold greater than that available to a normal coronavirus infection. The amount of virus present in the infected miners, sufficient to hospitalise all of them and kill half of them, was thus proportionately very large.
Evolutionary change is in large part a function of the population size. The lungs of the miners, we suggest, supported a very high viral load leading to proportionately rapid viral evolution.
Furthermore, according to the Master’s thesis, the immune systems of the miners were compromised and remained so even for those discharged. This weakness on the part of the miners may also have encouraged evolution of the virus.
iii) The length of infection experienced by the miners (especially patients 2, 3 and 4) far exceeded that of an ordinary coronavirus infection. From first becoming too sick to work in the mine, patient 2 survived 57 days until he died. Patient 3 survived 120 days after stopping work. Patient 4 survived 117 days and then was discharged as cured. Each had been exposed in the mine for 14 days prior to the onset of severe symptoms; thus each presumably had nascent infections for some time before calling in sick (See Table 2 of the thesis).
In contrast, in ordinary coronavirus infections the viral infection is cleared within about ten to fourteen days after being acquired (Tay et al., 2020). Thus, unlike most sufferers from coronavirus infection, the hospitalised miners had very long-term bouts of disease characterised by a continuous high load of virus. In the cases of patients 3 and 4 their illnesses lasted over 4 months.
iv) Coronaviruses are well known to recombine at very high rates: 10% of all progeny in a cell can be recombinants (Makino et al., 1986; Banner and Lai, 1991; Dudas and Rambaut, 2016). In normal virus evolution the mutation rate and the selection pressure are the main foci of attention. But in the case of a coronavirus adapting to a new host where many mutations distributed all over the genome are required to fully adapt to the new host, the recombination rate is likely to be highly influential in determining the overall speed of adaptation by the virus population (Baric et al., 1997).
Inside the miners a large tissue was simultaneously infected by a population of poorly-adapted viruses, with each therefore under pressure to adapt. Even if the starting population of virus lacked any diversity, many individual viruses would have acquired mutations independently but only recombination would have allowed these mutations to unite in the same genome. To recombine, viruses must be present in the same cell. In such a situation the particularities of lung tissues become potentially important because the existence of airways (bronchial tubes, etc.) allows partially-adapted viruses from independent viral populations to travel to distal parts of the lung (or even the other lung) and encounter other such partially-adapted viruses and populations. This movement around the lungs would likely have resulted in what amounted to a passaging effect without the need for a researcher to infect new tissues. Indeed, in the Master’s thesis the observation is several times made that areas of the lungs of a specific patient would appear to heal even while other parts of the lungs would become infected.
v) There were also a number of unusual things about the bat coronaviruses in the mine. They were abnormally abundant but also there were many different kinds, often causing co-infections of the bats (Ge et al., 2016). Viral co-infections are often more infectious or more pathogenic (Latham and Wilson, 2007).
As the WIV researchers remarked about the bats in the mine:
“we observed a high rate of co-infection with two coronavirus species and interspecies infection with the same coronavirus species within or across bat families. These phenomena may be owing to the diversity and high density of bat populations in the same cave, facilitating coronavirus intra- and interspecies transmissions, which may result in recombination and acceleration of coronavirus evolution.” (Ge et al., 2016).
The diversity of coronaviruses in the mine suggests that the miners were similarly exposed and that their illness may potentially have begun as co-infections.
Combining these observations, we propose that the miners’ lungs offered an unprecedented opportunity for accelerated evolution of a highly bat-adapted coronavirus into a highly human-adapted coronavirus and that decades of ordinary coronavirus evolution could easily have been condensed into months. However, we acknowledge that these conditions were unique. They and their scale have no exact scientific precedent we can refer to and they would be hard to replicate in a lab; thus it is important to emphasize that our proposal is fully consistent with the underlying principles of viral evolution as understood today.
In support of the MMP theory we also know something about the samples taken from the miners. According to the Master’s thesis, samples were taken from patients for “scientific research” and blood samples (at least) were sent to the WIV.
“In the later stage we worked with Dr. Zhong Nan Shan and did some sampling. The patient* tested positive for serum IgM by the WuHan Institute of Virology. It suggested the existence of virus infection” (p62 in the section “Comprehensive Analysis”.)
(*The original does not specify the number of patients tested.)
The Master’s thesis also states its regret that no samples for research were taken from patients 1 and 2, implying that samples were taken from all the others.
We further know that, on June 27th, 2012, the doctors performed an unexplained thymectomy on patient 4. The thymus is an immune organ that can potentially be removed without greatly harming the patient and it could have contained large quantities of virus. Beyond this the Master’s thesis is unfortunately unclear on the specifics of what sampling was done, for what purpose, and where each particular sample went.
Given the interests of the Shi lab in zoonotic origins of human disease, once such a sample was sent to them, it would have been obvious and straightforward for them to investigate how a virus from bats had managed to infect these miners. Any viruses recoverable from the miners would likely have been viewed by them as a unique natural experiment in human passaging offering unprecedented and otherwise-impossible-to-obtain insights into how bat coronaviruses can adapt to humans.
The logical course of such research would be to sequence viral RNA extracted directly from unfrozen tissue or blood samples and/or to generate live infectious clones for which it would be useful (if not imperative) to amplify the virus by placing it in human cell culture. Either technique could have led to accidental infection of a lab researcher.
Our supposition as to why there was a time lag between sample collection (in 2012/2013) and the COVID-19 outbreak is that the researchers were awaiting BSL-4 lab construction and certification, which was underway in 2013 but delayed until 2018.
We propose that, when frozen samples derived from the miners were eventually opened in the Wuhan lab they were already highly adapted to humans to an extent possibly not anticipated by the researchers. One small mistake or mechanical breakdown could have led directly to the first human infection in late 2019.
Thus, one of the miners, most likely patient 3, or patient 4 (whose thymus was removed), was effectively patient zero of the COVID-19 epidemic. In this scenario, COVID-19 is not an engineered virus; but, equally, if it had not been taken to Wuhan and no further molecular research had been performed or planned for it then the virus would have died out from natural causes, rather than escaped to initiate the COVID-19 pandemic.
Evidence in favour of the MMP proposal
Our proposal is consistent with all the principal undisputed facts concerning SARS-CoV-2 and its origin. The MMP proposal has the additional benefit of reconciling many observations concerning SARS-CoV-2 that have proven difficult to reconcile with any natural zoonotic hypothesis.
For instance, using different approaches, numerous researchers have concluded that the SARS-CoV-2 spike protein has a very high affinity for the human ACE2 receptor (Walls et al., 2020; Piplani et al., 2020; Shang and Ye et al., 2020; Wrapp et al., 2020). Such exceptional affinities, ten to twenty times as great as that of the original SARS virus, do not arise at random, making it very hard to explain in any other way than for the virus to have been strongly selected in the presence of a human ACE2 receptor (Piplani et al., 2020).
In addition to this, a recent report found that the spike of RaTG13 binds the human ACE2 receptor (Shang and Ye et al., 2020). We proposed above that the virus in the mine directly infected humans lung cells. The main determinant of cell infection and species specificity of coronaviruses is initial receptor binding (Perlman and Netland, 2009). Thus RaTG13, unlike most bat coronaviruses, probably can enter and infect human cells, providing biological plausibility to the idea that the miners became infected with a coronavirus resembling RaTG13.
Moreover, the receptor binding domain (RBD) of SARS-CoV-2, which is the region of the spike that physically contacts the human ACE2 receptor, has recently been crystallised to reveal its spatial structure (Shang and Ye et al., 2020). These authors found close structural similarities between the spikes of SARS-CoV-2 and RaTG13 in how they bound the human ACE2 receptor:
“Second, as with SARS-CoV-2, bat RaTG13 RBM [a region of the RBD] contains a similar four-residue motif in the ACE2 binding ridge, supporting the notion that SARS-CoV-2 may have evolved from RaTG13 or a RaTG13-related bat coronavirus (Extended Data Table 3 and Extended Data Fig. 7). Third, the L486F, Y493Q and D501N residue changes from RaTG13 to SARS CoV-2 enhance ACE2 recognition and may have facilitated the bat-to-human transmission of SARS-CoV-2 (Extended Data Table 3 and Extended Data Fig. 7). A lysine-to-asparagine mutation at the 479 position in the SARS-CoV RBD (corresponding to the 493 position in the SARS-CoV-2 RBD) enabled SARS-CoV to infect humans. Fourth, Leu455 contributes favourably to ACE2 recognition, and it is conserved between RaTG13 and SARS CoV-2; its presence in the SARS CoV-2 RBM may be important for the bat-to-human transmission of SARS-CoV-2 (Shang and Ye et al., 2020). (italics added)
The significance of this molecular similarity is very great. Coronaviruses have evolved a diverse set of molecular solutions to solve the problem of binding ACE2 (Perlman and Netland, 2009; Forni et al., 2017). The fact that RaTG13 and SARS CoV-2 share the same solution makes RaTG13 a highly likely direct ancestor of Sars-CoV-2.
A further widely noted feature of SARS-CoV-2 is its furin site (Coutard et al., 2020). This site is absent from RaTG13 and other closely related coronaviruses. The most closely related virus with such a site is the highly lethal MERS (which broke out in 2012). Possession of a furin site enables SARS-CoV-2 (like MERS) to infect lungs and many other body tissues (such as the gastrointestinal tract and neurons), explaining much of its lethality (Hoffman et al., 2020; Lamers et al., 2020). However, no convincing explanation for how SARS-CoV-2 acquired this site has yet been offered. Our suggestion is that it arose due to the high selection pressure which existed in the miner’s lungs and which in general worked to ensure that the virus became highly adapted to the lungs. This explanation, which encompasses how SARS-CoV-2 came to target lung tissues in general, is an important aspect of our proposal.
The implication is therefore that the furin site was not acquired by recombination with another coronavirus and simply represents convergent evolution (as suggested by Andersen et al., 2020).
An intriguing alternative possibility is that SARS-CoV-2 acquired its furin site directly from the miner’s lungs. Humans possess an epithelial sodium channel protein called ENaC-a whose furin cleavage site is identical over eight amino acids to SARS-CoV-2 (Anand et al., 2020). ENaC-a protein is present in the same airway epithelial and lung tissues infected by SARS-CoV-2. It is known from plants that positive-stranded RNA viruses recombine readily with host mRNAs (Greene and Allison, 1994; Greene and Allison, 1996; Lommel and Xiong, 1991; Borja et al., 2007). The same evidence base is not available for positive-stranded animal RNA viruses, (though see Gorbalenya, 1992) but if plant viruses are a guide then acquisition of its furin site via recombination with the mRNA which encodes ENaC-a by SARS-CoV-2 is a strong possibility.
A further feature of SARS-CoV-2 has been the very limited adaptive evolution of its genome since the pandemic began (Zhan et al., 2020; van Dorp et al., 2020; Starr et al., 2020). It is a well-established principle that viruses that jump species undergo accelerated evolutionary change in their new host (e.g. Baric et al., 1997). Thus, SARS and MERS (both coronaviruses) underwent rapid and readily detectable adaptation to their new human hosts (Forni et al., 2017; Dudas and Rambaut, 2016). Such an adaptation period has not been observed for SARS-CoV-2 even though it has now infected many more individuals than SARS or MERS did. This has even led to suggestions that the SARS-CoV-2 virus had a period of cryptic circulation in humans infections that predated the pandemic (Chaw et al., 2020). The sole mutation consistently observed to accumulate across multiple studies is a D614G substitution in the spike protein (e.g. Korber et al., 2020). The numerically largest analysis of SARS-CoV-2 genomes, however, found no evidence at all for adaptive evolution, even for D614G (van Dorp et al., 2020).
The general observation is therefore that Sars-CoV-2 has remained functionally unchanged or virtually so (except for inconsequential genetic changes) since the pandemic began. This is a very important observation. It implies that SARS-CoV-2 is highly adapted across its whole set of component proteins and not just at the spike (Zhan et al., 2020). That is to say, its evolutionary leap to humans was completed before the 2019 pandemic began.
It is hard to imagine an explanation for this high adaptiveness other than some kind of passaging in a human body (Zhan et al., 2020). Not even passaging in human cells could have achieved such an outcome.
Two examples illustrate this point. In a follow up to Shang and Ye et al., (2020), a similar group of Minnesota researchers identified a distinct strategy by which the spike (S) protein (which contains the receptor bind domain; RBD) of SARS-CoV-2 evades the human immune system (Shang and Wan et al., 2020). This strategy involves more effective hiding of its RBD, but it implies again that the spike and the RBD evolved in tandem and in the presence of the human immune system (i.e. in a human body and not in tissue culture).
The Andersen authors, in their critique of a possible engineered origin for SARS-CoV-2, also stress the need for passaging in whole humans:
“Finally, the generation of the predicted O-linked glycans is also unlikely to have occurred during cell-culture passage, as such features suggest the involvement of an immune system” (Andersen et al., 2020).
The final point that we would like to make is that the principal zoonotic origin thesis is the one proposed by Andersen et al. Apart from being poorly supported this thesis is very complex. It requires two species jumps, at least two recombination events between quite distantly related coronaviruses and the physical transfer of a pangolin (having a coronavirus infection) from outside China (Andersen et al., 2020). Even then it provides no logical explanation of the adaptedness of SARS-CoV-2 across its whole genome or why the virus emerged in Wuhan.
By contrast, our MMP proposal requires only the one species jump, which is documented in the Master’s thesis. Although we do not rule out a possible role for mixed infections in the lungs of the miners, nor the possibility of recombination between closely related variants in those lungs, nor the potential acquisition of the furin site from a host mRNA, only mutation was needed to derive SARS-CoV-2 from RaTG13. Hence our attention earlier to the figure from P. Zhou et al., 2020 showing that RaTG13 is the most closely related virus to SARS-CoV-2 over its entire length. This extended similarity is perfectly consistent with a mutational origin of SARS-CoV-2 from RaTG13.
In short, the MMP theory is a plausible and parsimonious explanation of all the key features of the COVID-19 pandemic and its origin. It accounts for the propensity of SARS-CoV-2 infections to target the lungs; the apparent preadapted nature of the virus; and its transmission from bats in Yunnan to humans in Wuhan.
Further questions
The hypothesis that SARS-CoV-2 evolved in the Mojiang miner’s lungs potentially resolves many scientific questions about the origin of the pandemic. But it raises others having to do with why this information has not come to light hitherto. The most obvious of these concern the actions of the Shi lab at the WIV.
Why did the Shi lab not acknowledge the miners’ deaths in any paper describing samples taken from the mine (Ge et al., 2016 and P. Zhou et al., 2020)? Why in the title of the Ge at al. 2016 paper did the Shi lab call it an “abandoned” mine? When they published the sequence of RaTG13 in Feb. 2020, why did the Shi lab provide a new name (RaTG13) for BtCoV/4991 when they had by then cited BtCoV/4991 twice in publications and once in a genome sequence database and when their sequences were from the same sample and 100% identical (P. Zhou et al., 2020)? If it was just a name change, why no acknowledgement of this in their 2020 paper describing RaTG13 (Bengston, 2020)? These strange and unscientific actions have obscured the origins of the closest viral relatives of SARS-CoV-2, viruses that are suspected to have caused a COVID-like illness in 2012 and which may be key to understanding not just the origin of the COVID-19 pandemic but the future behaviour of SARS-CoV-2.
These are not the only questionable actions associated with the provenance of samples from the mine. There were five scientific publications that very early in the pandemic reported whole genome sequences for SARS-CoV-2 (Chan et al., 2020; Chen et al., 2020; Wu et al., 2020; P. Zhou et al., 2020; Zhu et al., 2020). Despite three of them having experienced viral evolutionary biologists as authors (George Gao, Zheng-li Shi and Edward Holmes) only one of these (Chen et al., 2020) succeeded in identifying the most closely related viral sequence by far: BtCoV/4991 a viral sequence in the possession of the Shi lab at the WIV that differed from SARS-CoV-2 by just 5 nucleotides.
As we noted in our earlier article, the most important of the questions surrounding the origins of SARS-CoV-2 could potentially be resolved by a simple examination of the complete lab notebooks and biosafety records of relevant researchers at the WIV. Now that a credible and testable lab escape hypothesis exists this task becomes potentially much easier. This moment thus represents an opportune one to renew that call for an independent and transparent investigation of the WIV.
In requesting an investigation we are aware that no scientific institution anywhere has made a comparable request. We believe that this failure undermines public trust in a “scientific response” to the pandemic. Instead, the scientific establishment has labeled the lab escape theory a “rumor“, an “unverified theory” and a “conspiracy” when its proper name is a hypothesis. By taking this stance the scientific establishment has given the unambiguous message that scientists who take the possibility of a lab origin seriously are jeopardising their careers. Thus, while countless scientific publications on the pandemic assert in their introductions that a zoonotic origin for SARS-CoV-2 is a matter of fact or near-certainty (and Andersen et al has 860 citations as of July 14th), there is still not one published scientific paper asserting that a lab escape is even a credible hypothesis that deserves investigation.
Anyone who doubts this pressure should read the interview with Birger Sørensen in Norway’s Minerva magazine in which Sørensen discusses the “reluctance” of journals to publish his assessment that the existence of a virus that is “exceptionally well adjusted to infect humans” is “suspicious” and “cannot have evolved naturally”. The source of this reluctance, says Sørensen, is not rationality or scientific evidence. It results from conflicts of interest. This mirrors our experience. To find genuinely critical analysis of COVID-19 origin theories one has to go to Twitter, blog posts, and preprint servers. The malaise runs deep when even scientists start to complain that they don’t trust science.
We nevertheless hope that journalists will investigate some of the conflicts of interest that are keeping scientists and institutions from properly investigating the lab escape hypothesis.
References
Anand, P., Puranik, A., Aravamudan, M., Venkatakrishnan, A. J., & Soundararajan, V. (2020). SARS-CoV-2 strategically mimics proteolytic activation of human ENaC. Elife, 9, e58603.
Andersen, K. G., Rambaut, A., Lipkin, W. I., Holmes, E. C., & Garry, R. F. (2020). The proximal origin of SARS-CoV-2. Nature medicine, 26(4), 450-452.
Banner, L. R., & Mc Lai, M. (1991). Random nature of coronavirus RNA recombination in the absence of selection pressure. Virology, 185(1), 441-445.
Baric, R. S., Yount, B., Hensley, L., Peel, S. A., & Chen, W. A. N. (1997). Episodic evolution mediates interspecies transfer of a murine coronavirus. Journal of virology, 71(3), 1946-1955.
Becker, Y. (2000). Evolution of viruses by acquisition of cellular RNA or DNA nucleotide sequences and genes: an introduction. Virus Genes, 21(1-2), 7-12.
Bengston, D. (2020). All journal articles evaluating the origin or epidemiology of SARS-CoV-2 that utilize the RaTG13 bat strain genomics are potentially flawed and should be retracted. OSF Preprint: https://osf.io/wy89d
Borja, M., Rubio, T., Scholthof, H. B., & Jackson, A. O. (1999). Restoration of wild-type virus by double recombination of tombusvirus mutants with a host transgene. Molecular Plant-Microbe Interactions, 12(2), 153-162.
Chan, J. F. W., Kok, K. H., Zhu, Z., Chu, H., To, K. K. W., Yuan, S., & Yuen, K. Y. (2020). Genomic characterization of the 2019 novel human-pathogenic coronavirus isolated from a patient with atypical pneumonia after visiting Wuhan. Emerging microbes & infections, 9(1), 221-236.
Chaw, S. M., Tai, J. H., Chen, S. L., Hsieh, C. H., Chang, S. Y., Yeh, S. H., … & Wang, H. Y. (2020). The origin and underlying driving forces of the SARS-CoV-2 outbreak. Journal of biomedical science, 27(1), 1-12.
Chen, L., Liu, W., Zhang, Q., Xu, K., Ye, G., Wu, W., … & Mei, Y. (2020). RNA based mNGS approach identifies a novel human coronavirus from two individual pneumonia cases in 2019 Wuhan outbreak. Emerging microbes & infections, 9(1), 313-319.
Coutard, B., Valle, C., de Lamballerie, X., Canard, B., Seidah, N. G., & Decroly, E. (2020). The spike glycoprotein of the new coronavirus 2019-nCoV contains a furin-like cleavage site absent in CoV of the same clade. Antiviral research, 176, 104742.
Dudas, G., & Rambaut, A. (2016). MERS-CoV recombination: implications about the reservoir and potential for adaptation. Virus evolution, 2(1).
Duggal, A., Pinto, R., Rubenfeld, G., & Fowler, R. A. (2016). Global variability in reported mortality for critical illness during the 2009-10 influenza A (H1N1) pandemic: a systematic review and meta-regression to guide reporting of outcomes during disease outbreaks. PloS one, 11(5), e0155044.
Forni, D., Cagliani, R., Clerici, M., & Sironi, M. (2017). Molecular evolution of human coronavirus genomes. Trends in microbiology, 25(1), 35-48.
Furmanski, M. (2014). Laboratory Escapes and “Self-fulfilling prophecy” Epidemics. Report: Center for Arms Control and Nonproliferation. PDF available at: https://armscontrolcenter.org/wp-content/uploads/2016/02/Escaped-Viruses-final-2-17-14-copy.pdf
Ge, X. Y., Li, J. L., Yang, X. L., Chmura, A. A., Zhu, G., Epstein, J. H., … & Zhang, Y. J. (2013). Isolation and characterization of a bat SARS-like coronavirus that uses the ACE2 receptor. Nature, 503(7477), 535-538.
Ge, X. Y., Wang, N., Zhang, W., Hu, B., Li, B., Zhang, Y. Z., … & Wang, B. (2016). Coexistence of multiple coronaviruses in several bat colonies in an abandoned mineshaft. Virologica Sinica, 31(1), 31-40.
Gibbs, A. J., Armstrong, J. S., & Downie, J. C. (2009). From where did the 2009’swine-origin’influenza A virus (H1N1) emerge?. Virology journal, 6(1), 207.
Gorbalenya, A. E. (1992). Host-related sequences in RNA viral genomes. In Seminars in Virology (Vol. 3, pp. 359-359). HARCOURT BRACE JOVANOVICH.
Greene, A. E., & Allison, R. F. (1994). Recombination between viral RNA and transgenic plant transcripts. Science, 263(5152), 1423-1425.
Greene, A. E., & Allison, R. F. (1996). Deletions in the 3 untranslated region of cowpea chlorotic mottle virus transgene reduce recovery of recombinant viruses in transgenic plants. Virology, 225(1), 231-234.
Hasleton, P. S. (1972). The internal surface area of the adult human lung. Journal of anatomy, 112(Pt 3), 391.
Herfst, S., Schrauwen, E. J., Linster, M., Chutinimitkul, S., de Wit, E., Munster, V. J., … & Rimmelzwaan, G. F. (2012). Airborne transmission of influenza A/H5N1 virus between ferrets. science, 336(6088), 1534-1541.
Hoffmann, M., Kleine-Weber, H., & Pöhlmann, S. (2020). A multibasic cleavage site in the spike protein of SARS-CoV-2 is essential for infection of human lung cells. Molecular Cell.
Hu, B., Zeng, L. P., Yang, X. L., Ge, X. Y., Zhang, W., Li, B., … & Luo, D. S. (2017). Discovery of a rich gene pool of bat SARS-related coronaviruses provides new insights into the origin of SARS coronavirus. PLoS pathogens, 13(11), e1006698.
Huang, Canping (2016) Novel Virus Discovery in Bat and the Exploration of Receptor of Bat Coronavirus HKU9. PhD thesis (Original in Chinese). National Institute for Viral Disease Control and Prevention, Chinese Center for Disease Control and Prevention, June 2016. Accessed on July 15, 2020: https://eng.oversea.cnki.net/kcms/detail/detail.aspx?dbcode=CDFD&QueryID=4&CurRec=1&dbname=CDFDLAST2018&filename=1017118517.nh
Huang, C., Wang, Y., Li, X., Ren, L., Zhao, J., Hu, Y., … & Cheng, Z. (2020). Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. The lancet, 395(10223), 497-506.
Korber, B., Fischer, W., Gnanakaran, S. G., Yoon, H., Theiler, J., Abfalterer, W., … & Partridge, D. G. (2020). Spike mutation pipeline reveals the emergence of a more transmissible form of SARS-CoV-2. bioRxiv.
Lamers, M. M., Beumer, J., van der Vaart, J., Knoops, K., Puschhof, J., Breugem, T. I., … & van Donselaar, E. (2020). SARS-CoV-2 productively infects human gut enterocytes. Science.
Latham, J. R., & Wilson, A. K. (2008). Transcomplementation and synergism in plants: implications for viral transgenes?. Molecular Plant Pathology, 9(1), 85-103.
Latinne, A., Hu, B., Olival, K. J., Zhu, G., Zhang, L., Li, H., … & Li, B. (2020). Origin and cross-species transmission of bat coronaviruses in China. bioRxiv.
Lee, J., Hughes, T., Lee, M. H., Field, H., Rovie-Ryan, J. J., Sitam, F. T., … & Lasimbang, H. (2020). No evidence of coronaviruses or other potentially zoonotic viruses in Sunda pangolins (Manis javanica) entering the wildlife trade via Malaysia. bioRxiv.
Letko, M., Marzi, A., & Munster, V. (2020). Functional assessment of cell entry and receptor usage for SARS-CoV-2 and other lineage B betacoronaviruses. Nature microbiology, 5(4), 562-569.
Li, W., Shi, Z., Yu, M., Ren, W., Smith, C., Epstein, J. H., … & Zhang, J. (2005). Bats are natural reservoirs of SARS-like coronaviruses. Science, 310(5748), 676-679.
Lipsitch, M. (2018). Why Do Exceptionally Dangerous Gain-of-Function Experiments in Influenza?. In Influenza Virus (pp. 589-608). Humana Press, New York, NY.
Lipsitch, M., & Galvani, A. P. (2014). Ethical alternatives to experiments with novel potential pandemic pathogens. PLoS Med, 11(5), e1001646.
Lommel, A., & Xiong, Z. (1991). Reconstitution of a functional red clover necrotic mosaic virus by recombinational rescue of the cell-to-cell movement gene expressed in a transgenic plant. Journal of Cellular Biochemistry A, 15, 151.
Lu, R., Zhao, X., Li, J., Niu, P., Yang, B., Wu, H., … & Bi, Y. (2020). Genomic characterisation and epidemiology of 2019 novel coronavirus: implications for virus origins and receptor binding. The Lancet, 395(10224), 565-574.
Makino, S. H. I. N. J. I., Keck, J. G., Stohlman, S. A., & Lai, M. M. (1986). High-frequency RNA recombination of murine coronaviruses. Journal of Virology, 57(3), 729-737.
McKinsey, D. S., & McKinsey, J. P. (2011, December). Pulmonary histoplasmosis. In Seminars in respiratory and critical care medicine (Vol. 32, No. 06, pp. 735-744). © Thieme Medical Publishers.
Menachery, V. D., Yount, B. L., Debbink, K., Agnihothram, S., Gralinski, L. E., Plante, J. A., … & Randell, S. H. (2015). A SARS-like cluster of circulating bat coronaviruses shows potential for human emergence. Nature medicine, 21(12), 1508-1513.
Pan, B., Chen, M., Pan, W., & Liao, W. (2013). Histoplasmosis: a new endemic fungal infection in China? Review and analysis of cases. Mycoses, 56(3), 212-221.
Perlman, S., & Netland, J. (2009). Coronaviruses post-SARS: update on replication and pathogenesis. Nature reviews microbiology, 7(6), 439-450.
Piplani, S., Singh, P. K., Winkler, D. A., & Petrovsky, N. (2020). In silico comparison of spike protein-ACE2 binding affinities across species; significance for the possible origin of the SARS-CoV-2 virus. arXiv preprint arXiv:2005.06199.
Rahalkar, M.C.; Bahulikar, R.A. Understanding the Origin of ‘BatCoVRaTG13’, a Virus Closest to SARS-CoV-2. Preprints 2020, 2020050322 https://www.preprints.org/manuscript/202005.0322/v2
Segreto, R., & Deigin, Y. Is considering a genetic-manipulation origin for SARS-CoV-2 a conspiracy theory that must be censored?. https://www.researchgate.net/profile/Rossana_Segreto/publication/340924249_Is_considering_a_genetic-manipulation_origin_for_SARS-CoV-2_a_conspiracy_theory_that_must_be_censored/links/5ed7c17992851c9c5e74f7dc/Is-considering-a-genetic-manipulation-origin-f
Shang, J., Wan, Y., Luo, C., Ye, G., Geng, Q., Auerbach, A., & Li, F. (2020). Cell entry mechanisms of SARS-CoV-2. Proceedings of the National Academy of Sciences, 117(21), 11727-11734.
Shang, J., Ye, G., Shi, K., Wan, Y., Luo, C., Aihara, H., … & Li, F. (2020). Structural basis of receptor recognition by SARS-CoV-2. Nature, 581(7807), 221-224.
Simonsen, L., Spreeuwenberg, P., Lustig, R., Taylor, R. J., Fleming, D. M., Kroneman, M., … & Paget, W. J. (2013). Global mortality estimates for the 2009 Influenza Pandemic from the GLaMOR project: a modeling study. PLoS Med, 10(11), e1001558.
Starr, T. N., Greaney, A. J., Hilton, S. K., Crawford, K. H., Navarro, M. J., Bowen, J. E., … & Bloom, J. D. (2020). Deep mutational scanning of SARS-CoV-2 receptor binding domain reveals constraints on folding and ACE2 binding. BioRxiv.
Tay, M. Z., Poh, C. M., Rénia, L., MacAry, P. A., & Ng, L. F. (2020). The trinity of COVID-19: immunity, inflammation and intervention. Nature Reviews Immunology, 1-12.
van Dorp, L., Richard, D., Tan, C. C., Shaw, L. P., Acman, M., & Balloux, F. (2020). No evidence for increased transmissibility from recurrent mutations in SARS-CoV-2. bioRxiv. doi: https://doi.org/10.1101/2020.05.21.108506
https://www.biorxiv.org/content/10.1101/2020.05.21.108506v1.abstract
Wadman, M., Couzin-Frankel, J., Kaiser, J., & Matacic, C. (2020). A rampage through the body. Science, 368(6489), 356-360.
Walls, A. C., Park, Y. J., Tortorici, M. A., Wall, A., McGuire, A. T., & Veesler, D. (2020). Structure, function, and antigenicity of the SARS-CoV-2 spike glycoprotein. Cell, 180, 281-292.
Wang, N., Luo, C., Liu, H., Yang, X., Hu, B., Zhang, W., … & Peng, C. (2019). Characterization of a new member of alphacoronavirus with unique genomic features in rhinolophus bats. Viruses, 11(4), 379.
Weiss, S., Yitzhaki, S., & Shapira, S. C. (2015). Lessons to be Learned from Recent Biosafety Incidents in the United States. The Israel Medical Association Journal: IMAJ, 17(5), 269-273.
Wrapp, D., Wang, N., Corbett, K. S., Goldsmith, J. A., Hsieh, C. L., Abiona, O., … & McLellan, J. S. (2020). Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science, 367(6483), 1260-1263.
Wu, Z., Yang, L., Yang, F., Ren, X., Jiang, J., Dong, J., … & Jin, Q. (2014). Novel henipa-like virus, Mojiang paramyxovirus, in rats, China, 2012. Emerging infectious diseases, 20(6), 1064.
Wu, F., Zhao, S., Yu, B., Chen, Y. M., Wang, W., Song, Z. G., … & Yuan, M. L. (2020). A new coronavirus associated with human respiratory disease in China. Nature, 579(7798), 265-269.
Xu, Li (2013) The analysis of 6 patients with severe pneumonia caused by unknown viruses. MSc thesis (Original in Chinese). Emergency Department and EICU, The 1st Affiliated Hospital of Kunming Medical University, Kunming. Accessed on July, 15, 2020: http://eng.oversea.cnki.net/Kcms/detail/detail.aspx?filename=1013327523.nh&dbcode=CMFD&dbname=CMF D2014
Zhan, S. H., Deverman, B. E., & Chan, Y. A. (2020). SARS-CoV-2 is well adapted for humans. What does this mean for re-emergence?. bioRxiv. doi: https://doi.org/10.1101/2020.05.01.073262
Zhang, L., Jackson, C. B., Mou, H., Ojha, A., Rangarajan, E. S., Izard, T., … & Choe, H. (2020). The D614G mutation in the SARS-CoV-2 spike protein reduces S1 shedding and increases infectivity. bioRxiv
Zhang, Y. Z., & Holmes, E. C. (2020). A genomic perspective on the origin and emergence of SARS-CoV-2. Cell.
Zhou, H., Chen, X., Hu, T., Li, J., Song, H., Liu, Y., … & Shi, W. (2020). A novel bat coronavirus reveals natural insertions at the S1/S2 cleavage site of the Spike protein and a possible recombinant origin of HCoV-19. bioRxiv.
Zhou, P., Yang, X. L., Wang, X. G., Hu, B., Zhang, L., Zhang, W., … & Chen, H. D. (2020). A pneumonia outbreak associated with a new coronavirus of probable bat origin. nature, 579(7798), 270-273.
Zhou, M., Zhang, X., & Qu, J. (2020). Coronavirus disease 2019 (COVID-19): a clinical update. Frontiers of medicine, 1-10.
Zhu, N., Zhang, D., Wang, W., Li, X., Yang, B., Song, J., … & Niu, P. (2020). A novel coronavirus from patients with pneumonia in China, 2019. New England Journal of Medicine.
 Infoblox: Dancing With Scammers - The Telegram Tango Investigation
Infoblox: Dancing With Scammers - The Telegram Tango Investigation Consumer NZ: This Mother’s Day, Give The Gift Of Scam Protection And Digital Confidence
Consumer NZ: This Mother’s Day, Give The Gift Of Scam Protection And Digital Confidence NZ Airports Association: Airlines And Airports Back Visa Simplification
NZ Airports Association: Airlines And Airports Back Visa Simplification Netsafe: Statement From Netsafe About Proposed Social Media Ban
Netsafe: Statement From Netsafe About Proposed Social Media Ban The Reserve Bank of New Zealand: 2024 General Insurance Stress Test Results Published Today
The Reserve Bank of New Zealand: 2024 General Insurance Stress Test Results Published Today  Worldline: School Holidays And Long Weekends Change Regional Spending Patterns In April
Worldline: School Holidays And Long Weekends Change Regional Spending Patterns In April